Вступление
20 декабря 2023 г. закончилось соревнование CAFA5: активная фаза длилась с середины апреля по конец августа, затем ждали результатов на честном тестовом наборе данных, собранном после окончания приема сабмитов. Маленькая команда R-щиков в составе Антонины Долгоруковой и меня выступила достаточно успешно, заняв 16 место на публичном лидерборде и 13 на приватном, буквально на десятитысячных долях (kaggle-style) дотянув до золотой медали. Можно ознакомиться с кратким описанием решения, ну а в этом сообщении рассказывается максимально подробно, что делали в ходе соревнования, чему научились и на чем обломались.
Описание соревнования
Critical Assessment of Functional Annotation 5, как можно догадаться, является уже пятым соревнованием из этой серии. На платформе kaggle проводится впервые.
Цель состоит в том, чтобы научиться предсказывать новые функциональные свойства белков, которые описываются GO-аннотациями. Аннотация состоит из названия “генного продукта” (в нашем случае белка), собственно GO-терма (например, GO:0016887 обозначает наличие каталитической активности для реакции гидролиза АТФ), ссылки на источник аннотации и кодового обозначения доказательного уровня этой аннотации. Термы образуют иерархическую структуру (направленный ациклический граф), называемую онтологией. Важно, что структура эта не является древовидной - см. картинку ниже. Понятия терм и аннотация далее будем использовать как синонимы.
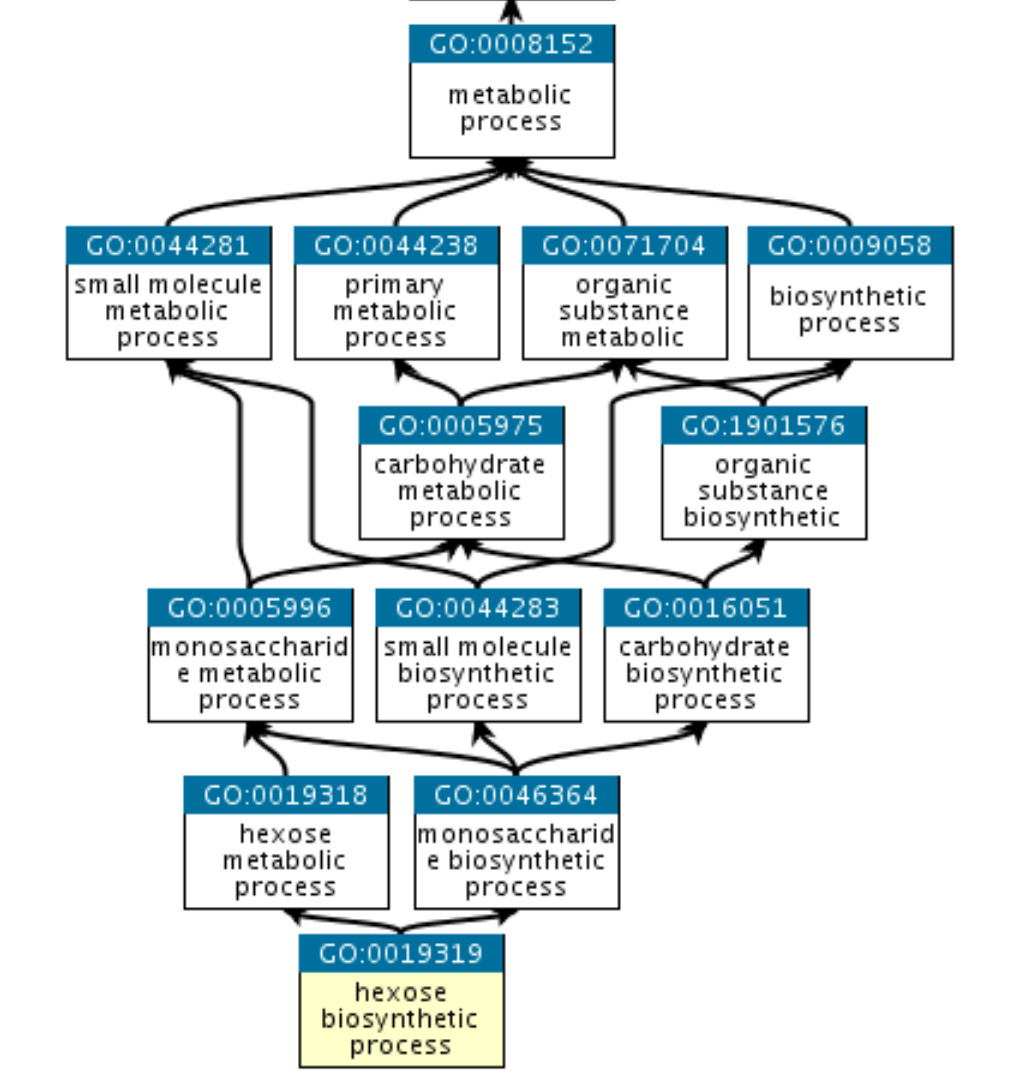
Вся онтология делится на 3 субонтологии:
Molecular Function (MF) - молекулярная функция;
Biological Process (BP) - биологический процесс;
Cellular Component (CC) - компонент клетки.
Разумеется, онтология является динамической структурой и постоянно обновляется по мере появления новых знаний. Речь идет не о разметке новых белков известными термами, а о появлении или удалении термов и изменении связей между ними. Организаторы предоставили фиксированную на весь период соревнования версию онтологии от 1 января 2023 года в виде OBO-файла.
Качество предсказания оценивается отдельно для субонтологий. В каждой оценке участвуют белки, которые 1) не имели экспериментальных аннотаций в данной субонтологии на момент окончания приема сабмитов в соревновании и 2) получили экспериментальные аннотации за период с момента окончания приема сабмитов и до окончания соревнования. Отсюда следуют две важные особенности соревнования, а именно отсутствие наперед заданного тестового набора белков (есть лишь так называемый “тестовый суперсет”, который к тому же пересекается с обучающим набором) и необходимости максимально полно предсказывать все аннотации для всех белков.
Разметка была представлена экспериментальными аннотациями из базы данных UniProtKB от 17 ноября 2022 года. Допускалось использование более свежих данных с юнипрота, а также с других публичных ресурсов, таких как QuickGO. Забегая вперед: в одной из моделей мы использовали как раз такие обновленные данные. Особенность разметки в том, что она фундаментально неполная, имеется множество ложноотрицательных значений (отсутствие аннотации не означает отсутствия соответствующей функции у белка). Соответственно, чисто по постановке это positive-unlabeled learning - задача обучения на положительных и неразмеченных примерах. Но все решали задачу как многоклассовую классикацию с несколькими метками (multilabel).
Признакового описания как такового организаторы не предоставили. Были даны лишь белковые сиквенсы, для которых участники соревнования получали векторные представления (эмбеддинги) при помощи предварительно обученных языковых моделей. Мы в основном использовали эмбеддинги ESM-2, которыми поделился andreylalaley
Метрика качества
Метрикой качества была взвешенная F1-мера: каждый предсказываемый терм имеет свой вес, предсказания должны быть в диапазоне от 0 до 1, метрика вычисляется для всех возможных пороговых значений с шагом 0.001 (отдельно по субонтологиям), затем берутся максимальные значения по субонтологиям и усредняются. При этом, если для белка отсутствует предсказание какого-то родительского терма из субонтологии, но есть предсказания его дочерних термов, максимум предсказаний дочерних термов становится предсказанием этого родительского терма.
Если вам кажется, что это довольно сложно, то вам не кажется. Сами организаторы допустили ошибку в коде для расчета метрики, и в течение половины соревнования результаты на лидерборде были неправильными. Код был выложен на гитхабе, а ошибку обнаружил участник из команды, занявшей в итоге 2 место. Мы же изначально не реализовали расчет этой метрики для локальной валидации и даже не изучили код от организаторов. Вместо этого ориентировались на ROC-AUC, что, как назло, неплохо коррелировало с результатом на лидерборде и позволило подняться до 5 места. А потом при пересчете метрики по правильной формуле улетели на 200+ место, и всю работу нужно было начинать заново. Так что это первая ошибка, существенно повлиявшая на итоговый результат.
Для расчета метрики нужна таблица следующего вида:
Подготовка таблицы с предиктами
library(data.table)
library(qs)
library(future)
library(furrr)
library(knitr)
future::plan("multisession", workers = 10)
preds <- qread("valid_top_n/valid_top_n_corr.qs") # prediction
test_response <- qread("valid_top_n/test_response.qs") # ground truth
test_response[, truth := 1]
preds <- merge(preds, test_response, by = c("EntryID", "term"), all = TRUE)
preds[unique(test_response[, .(term, aspect)]),
aspect := i.aspect, on = "term"]
preds[is.na(pred), pred := 0]
preds[is.na(truth), truth := 0]
term_weights <- fread("data_source/IA.txt")
setnames(term_weights, names(term_weights), c("term", "weight"))
preds[term_weights, weight := i.weight, on = "term"]
kable(head(preds))| EntryID | term | pred | aspect | truth | weight |
|---|---|---|---|---|---|
| A0A024CBD6 | GO:0000003 | 0.178 | BPO | 0 | 3.439404 |
| A0A024CBD6 | GO:0001669 | 0.043 | CCO | 0 | 2.509487 |
| A0A024CBD6 | GO:0001775 | 0.051 | BPO | 0 | 4.051549 |
| A0A024CBD6 | GO:0001817 | 0.108 | BPO | 0 | 1.325809 |
| A0A024CBD6 | GO:0002020 | 0.024 | MFO | 0 | 4.032960 |
| A0A024CBD6 | GO:0002376 | 0.330 | BPO | 0 | 4.490188 |
По каждой субонтологии рассчитываем взвешенные точность и полноту для каждого порогового значения с шагом 0.01 (на самом деле метрика учитывается с точностью до 0.001, но так можно значительно ускорить вычисления, а результат почти не изменится). Затем считаем F1-меру для каждого порогового значения и находим максимум.
Расчет максимальной взвешенной F1-меры
# Weighted precision
wpr <- function(tau = 0.1, preds) {
dt <- preds[,
.(EntryID, term, pred = ifelse(pred >= tau, 1, 0),
truth, weight)
]
# All proteins with at least one predicted term
dt <- dt[, .SD[any(pred == 1)], by = EntryID]
dt[,
.SD[pred == 1 & truth == 1, sum(weight)] / .SD[pred == 1, sum(weight)],
by = EntryID
][, mean(V1, na.rm = TRUE)]
}
# Weighted recall
wrc <- function(tau = 0.1, preds) {
dt <- preds[,
.(EntryID, term, pred = ifelse(pred >= tau, 1, 0),
truth, weight)
]
dt[,
.SD[pred == 1 & truth == 1, sum(weight)] / .SD[truth == 1, sum(weight)],
by = EntryID
][, mean(V1, na.rm = TRUE)]
}
f1_score <- function(dt) {
dt_pr <- data.table(
tau = seq(1, 0.000, by = -0.01)
)
dt_pr[, recall := future_map_dbl(tau, wrc, preds = dt,
.progress = TRUE)]
dt_pr[, precision := future_map_dbl(tau, wpr, preds = dt,
.progress = TRUE)]
dt_pr[, f1 := 2 * precision * recall / (precision + recall)]
dt_pr[, round(max(f1, na.rm = TRUE), 4)]
}
preds[
sample(1:preds[, .N], 1000), # Small sample for speed up
.(max_f1 = f1_score(.SD)),
by = aspect
] aspect max_f1
1: BPO 0.3696
2: CCO 0.6667
3: MFO 0.5854Даже при использовании 10 и более потоков на реальных данных эта метрика качества считается довольно долго, что также ограничивало нас в ее использовании для валидации. Возможно, имело смысл поискать пути дальнейшей оптимизации вычислений, да и просто проверить пусть меньше гипотез, зато по более правильной метрике.
Также среди опубликованных решений был обнаружен интересный вариант функции потерь - F1 loss. Эта функция является дифференцируемой, в отличие от F1-меры. Ее использование и как функции потерь, и как метрики качества вполне могло улучшить результат и/или сократить время вычислений.
Подготовка данных
Как было сказано выше, в качестве признаков при обучении моделей использовались эмбеддинги белков, полученные при помощи языковых моделей. Мы остановились на модели ESM-2 с 3 млрд. параметров, которая на выходе дает эмбеддинги аминокислотных остатков размерности 2560 (per-residue embeddings). Эмбеддинги белков получались простым усреднением. Альтернативой является обучение непосредственно на аминокислотных эмбеддингах с использованием сверток/аттеншенов, но этот вариант, насколько нам известно, ни у кого не взлетел.
Готовые эмбеддинги в формате .npy взяли здесь и пересохранили для удобства в qs:
Сохранение эмбеддингов в формате qs
library(data.table)
library(qs)
library(reticulate)
use_condaenv("tf")
np <- import("numpy")
# Train dataset with esm2_t36_3B embeddings ------------------------------------
train_ids <- np$load("esm2_t36_3B_embeds/train_ids_esm2_t36_3B_UR50D.npy")
train_embeds <- np$load("esm2_t36_3B_embeds/train_embeds_esm2_t36_3B_UR50D.npy")
train_features <- data.table(EntryID = train_ids)
cols <- paste0("f", 1:ncol(train_embeds))
train_features[, (cols) := as.data.table(train_embeds)]
# Sort by EntryID
setkey(train_features, "EntryID")
qsave(train_features, "data_prepared/train_features_esm2_t36_3B.qs")
# Test dataset with esm2_t36_3B embeddings -------------------------------------
test_ids <- np$load("esm2_t36_3B_embeds/test_ids_esm2_t36_3B_UR50D.npy")
test_embeds <- np$load("esm2_t36_3B_embeds/test_embeds_esm2_t36_3B_UR50D.npy")
test_features <- data.table(EntryID = test_ids)
cols <- paste0("f", 1:ncol(test_embeds))
# Old version with wrong shape
# test_features[, (cols) := as.data.table(test_embeds[1:length(test_ids), ])]
test_features[, (cols) := as.data.table(test_embeds)]
# Remove duplicates
test_features <- test_features[, .SD[1], by = EntryID]
# Sort by EntryID
setkey(test_features, "EntryID")
qsave(test_features, "data_prepared/test_features_esm2_t36_3B.qs")Модель имеет ограничение в 1022 аминокислотных остатка, более длинные последовательности просто обрезались. Были некоторые сомнения в правильности такого решения, поэтому в порядке эксперимента получили полные эмбеддинги для всех белков - см. ноутбук esm2_correct_embeds (на каггле запустить не получится, нужна видеокарта c 24 Гб памяти). Алгоритм следующий:
У нас есть белок из
seq_len=2500а.к., в сеть влазитmax_allowed_len=1022а.к. Запихиваем первые 1022 а.к., получаем аминокислотные эмбеддинги размерности 1022х2560.Выбрасываем первые
min(seq_len-max_allowed_len, max_allowed_len/2)а.к. В данном примере выбрасываем 511, если бы в белке было 1024 а.к., выбросили бы первые 2.Из оставшейся последовательности берем снова первые 1022 а.к., получаем эмбеддинги размерности 1022х2560. В них нас будут интересовать последние
min(seq_len-max_allowed_len, max_allowed_len/2)шт., то есть те, которых не было в прошлом предикте.Повторяем пункты 3-4, пока не получим эмбеддинги всех аминокислотных остатков, затем усредняем.
Перекрывающиеся кусочки последовательности нужны для сохранения контекста.
Этот вариант не дал прироста качества, поэтому попробовали также использовать эмбеддинги для последовательностей, обрезанных до 900 а.к. (это тоже ничего не дало). Код в таком случае упрощается:
Получение эмбеддингов ESM-2 для макс. длины 900
max_allowed_len = 900
embed_layer = 36
for i, (id, seq) in enumerate(zip(list_ids, list_seqs)):
if i % 100 == 0:
print(i,'sequences processed')
# Embeds of first max_allowed_len residuals
data = [(id, seq[0:min(len(seq), max_allowed_len)])]
batch_labels, batch_strs, batch_tokens = batch_converter(data)
batch_tokens = batch_tokens.cuda()
tokens_len = (batch_tokens != alphabet.padding_idx).sum(1)
with torch.no_grad():
results = model(batch_tokens,
repr_layers=[embed_layer],
return_contacts=False)
token_representations = results["representations"][embed_layer]
token_representations = token_representations[:, 1:(tokens_len-1), :]
token_representations = token_representations.squeeze(dim=0)
protein_embed = token_representations.mean(0)
protein_embed = protein_embed.cpu().numpy()
np.save(f"train_embeds/{id}.npy", protein_embed) Во всех случаях эмбеддинги каждого белка сохранялись в отдельном файле. Это медленнее, чем сразу формировать одну большую матрицу в ОЗУ, зато позволяет не потерять результаты вычислений, если что-то пошло не так.
Полученные эмбеддинги также пересохранили в qs:
Сохранение обновленных эмбеддингов в формате qs
if (!dir.exists("data_prepared")) dir.create("data_prepared")
library(data.table)
library(qs)
library(reticulate)
use_condaenv("tf")
np <- import("numpy")
# TRAIN -----------------------------------------------------------------------
ids <- tools::file_path_sans_ext(
list.files("esm2_t36_3B_embeds/train_embeds_long_fixed/")
)
files <- list.files("esm2_t36_3B_embeds/train_embeds_long_fixed/",
full.names = TRUE)
embeds <- sapply(files, np$load)
embeds <- t(embeds)
rownames(embeds) <- ids
colnames(embeds) <- paste0("f", 1:ncol(embeds))
embeds <- as.data.table(embeds, keep.rownames = "EntryID")
embeds <- unique(embeds)
qsave(embeds, "data_prepared/train_features_esm2_t36_3B_long_fixed.qs")
# replace embeds in train_features_esm2_t36_3B
train_features <- qread("data_prepared/train_features_esm2_t36_3B.qs")
cols <- paste0("f", 1:ncol(train_features[, !"EntryID"]))
train_features[embeds, (cols) := mget(sprintf("i.%s", cols)), on = "EntryID"]
qsave(train_features, "data_prepared/train_features_esm2_t36_3B_fixed.qs")
# TEST -------------------------------------------------------------------------
ids <- tools::file_path_sans_ext(
list.files("esm2_t36_3B_embeds/test_embeds_long_fixed/")
)
files <- list.files("esm2_t36_3B_embeds/test_embeds_long_fixed/",
full.names = TRUE)
embeds <- sapply(files, np$load)
embeds <- t(embeds)
rownames(embeds) <- ids
colnames(embeds) <- paste0("f", 1:ncol(embeds))
embeds <- as.data.table(embeds, keep.rownames = "EntryID")
embeds <- unique(embeds)
qsave(embeds, "data_prepared/test_features_esm2_t36_3B_long_fixed.qs")
# replace embeds in test_features_esm2_t36_3B
test_features <- qread("data_prepared/test_features_esm2_t36_3B.qs")
cols <- paste0("f", 1:ncol(test_features[, !"EntryID"]))
test_features[embeds, (cols) := mget(sprintf("i.%s", cols)), on = "EntryID"]
qsave(test_features, "data_prepared/test_features_esm2_t36_3B_fixed.qs")Затем была попытка получить эмбеддинги самой большой модели семейства ESM-2 на 15 млрд. параметров, для чего был арендован сервер с RTX A6000 на vast.ai. Но замеры качества на выборке из 20 000 белков показали, что такие эмбеддинги работают хуже тех, которые у нас уже были. Поэтому это направление забросили, и правильно сделали.
Что касается подготовки таргетов, то она осуществлялась из исходных файлов “на лету” в процессе обучения моделей. Поскольку термов огромное количество (тысячи), и многими из них аннотировано небольшое количество белков (часто всего 1), мы учились предсказывать только самые часто встречающиеся термы. Отбор для разных моделей проводили двумя способами: либо брали топ500 термов в каждой субонтологии (также проверялись другие количества), либо отбирали топ по минимальной частоте встречаемости (BP > 250, MF > 50 и CC > 50 - вариант, найденный в литеруре, и несколько других вариантов). Точный список моделей с разными наборами предсказываемых таргетов не принципиален; важно, что предсказания нескольких таких моделей, имеющих почти одинаковое качество, при усреднении дают хороший прирост этого самого качества.
При том или ином отборе топовых термов неизбежно оказывается, что часть белков в обучающей выборке не имеют ни одной аннотации выбранными термами. Удаление таких белков давало стабильный прирост качества предсказаний, более того, одна из моделей в итоговом ансамбле была обучена с дополнительным удалением 1000 белков, имеющих наименьшее количество аннотаций. Логика удаления слабо аннотированных белков и использования термов с не слишком маленьким количеством примеров обусловлена как борьбой с дисбалансом классов, так и попытками уменьшить долю ложноотрицательной разметки.
Обучение моделей
При подборе гиперпараметров для валидации использовали случайную выборку белков (либо из всех, либо из тех белков обучающей выборки, которые не пересекаются с тестовой). Альтернативные варианты - случайная выборка аннотаций по всем белкам или хронологическая выборка (валидация на более новых аннотациях) - показали себя не очень хорошо. Кросс-валидация с разбивкой на 5 или 10 фолдов была признана слишком вычислительно затратной.
Все лучшие модели были относительно простыми MLP вплоть до сетки, имеющей всего 1 скрытый слой. Ниже представлен полный скрипт для обучения чуть более сложной сетки с дропаутом. Для получения более стабильного решения данные разбивались на 10 фолдов и обучалось 10 моделей, каждая на 9 фолдах с использованием оставшегося фолда для ранней остановки. Затем предикты моделей усреднялись.
Обучение модели с использованием torch
if (!dir.exists("submits")) dir.create("submits")
library(torch)
library(luz)
library(data.table)
library(qs)
library(seqinr)
min_freq <- c("BPO" = 275, "CCO" = 55, "MFO" = 55)
train_sequences_file <- "data_source/Train/train_sequences.fasta"
train_terms_file <- "data_source/Train/train_terms.tsv"
test_features <- qread("data_prepared/test_features_esm2_t36_3B.qs")
#--------------------- Prepare terms by sequence data --------------------------
# fasta_seq <- qread(train_sequences_file)
fasta_seq <- read.fasta(
train_sequences_file,
seqtype = "AA",
as.string = TRUE
)
fasta_seq <- data.table(
EntryID = names(fasta_seq),
fasta = unlist(fasta_seq)
)
# Load train targets
train_targets_by_seq <- fread(train_terms_file , sep = '\t')
# Remove subontologies roots
train_targets_by_seq <- train_targets_by_seq[
! term %chin% c("GO:0008150", "GO:0005575", "GO:0003674")
]
# Add sequences
train_targets_by_seq[fasta_seq, sequence := i.fasta, on = "EntryID"]
# Replace duplicated EntryIDs with first one for each sequence
train_targets_by_seq[, EntryID := .SD[1, EntryID], by = sequence]
# Remove duplicated annotations
train_targets_by_seq <- unique(train_targets_by_seq)
uniqueN(train_targets_by_seq$EntryID)
#------------------ Predict by ontology ----------------------------------------
t0 <- Sys.time()
all_preds <- NULL
for (aspect_name in c("BPO", "CCO", "MFO") ) {
freq <- min_freq[aspect_name]
train_all_targets <- train_targets_by_seq[aspect == aspect_name]
labels_to_consider <- train_all_targets[, .(n = .N), by = term]
labels_to_consider <- labels_to_consider[, .SD[order(-n)]]
labels_to_consider <- labels_to_consider[n > freq]
train_targets <- dcast(
train_all_targets[labels_to_consider, on = "term", nomatch = 0],
EntryID ~ term, length,
fill = 0
)
cols <- setdiff(names(train_targets), "EntryID")
train_targets[, (cols) := lapply(.SD, as.numeric), .SDcols = cols]
train_features <- qread("data_prepared/train_features_esm2_t36_3B.qs")
# Remove proteins without selected terms
train_features <- train_features[
train_targets[, EntryID],
on = "EntryID",
nomatch = 0
]
# 10-fold cross-validation
set.seed(42)
cv10 <- data.table(id = 1:train_features[, .N])
cv10[, folds := sample(1:10, .N, replace = TRUE)]
n_features <- ncol(train_features) - 1
n_targets <- ncol(train_targets) - 1
d_hid1 <- 2048
wd = 0.25
lr = 0.01
bs = 256
dropout_prob = 0.5
num_epochs = 100
preds_cv <- data.table(
EntryID = rep(test_features$EntryID, times = n_targets),
term = rep(names(train_targets[, !"EntryID"]), each = test_features[, .N]),
pred = 0
)
mlp_fit_predict_torch <- function(fold, num_epochs = 1) {
cat("\n--------------- ", aspect_name, " ", train_features[, .N],
" proteins; Fold number: ", fold, " ---------------\n", sep = "")
train_ids <- cv10[folds != fold, id]
valid_ids <- cv10[folds == fold, id]
#------------Standardization (Z-score normalization)-----------------------#
train_tensor <- torch_tensor(as.matrix(train_features[train_ids, !"EntryID"]))
train_f <- torch_tensor(as.matrix(train_features[, !"EntryID"]))
test_f <- torch_tensor(as.matrix(test_features[, !"EntryID"]))
means <- torch_mean(train_tensor, dim = 1)
sds <- torch_std(train_tensor, dim = 1)
train_f <- (train_f - means) / sds
test_f <- (test_f - means) / sds
#--------------------------------------------------------------------------#
train_ds <- tensor_dataset(
train_f[train_ids, ],
torch_tensor(as.matrix(train_targets[train_ids, !"EntryID"]))
)
valid_ds <- tensor_dataset(
train_f[valid_ids, ],
torch_tensor(as.matrix(train_targets[valid_ids, !"EntryID"]))
)
test_ds <- tensor_dataset(
test_f
)
train_dl <- dataloader(train_ds, batch_size = bs, shuffle = TRUE)
valid_dl <- dataloader(valid_ds, batch_size = bs)
test_dl <- dataloader(test_ds, batch_size = bs)
#---------------------nn_module--------------------------------------------#
net <- nn_module(
initialize = function(n_features, n_targets, d_hid1) {
self$net <- nn_sequential(
nn_linear(n_features, d_hid1),
nn_relu(),
nn_dropout(dropout_prob),
nn_linear(d_hid1, d_hid1),
nn_relu(),
nn_dropout(dropout_prob),
nn_linear(d_hid1, n_targets),
nn_sigmoid()
)
},
forward = function(x) {
self$net(x)
}
)
fitted <- net %>%
setup(
loss = nn_bce_loss(),
optimizer = optim_adamw,
metrics = list(luz_metric_binary_auroc())
) %>%
set_hparams(
n_features = n_features,
n_targets = n_targets,
d_hid1 = d_hid1
) %>%
set_opt_hparams(weight_decay = wd) %>%
fit(
train_dl,
epochs = num_epochs,
valid_data = valid_dl,
callbacks = list(
luz_callback_lr_scheduler(
lr_one_cycle,
max_lr = lr,
epochs = num_epochs,
steps_per_epoch = length(train_dl),
call_on = "on_batch_end"
),
luz_callback_early_stopping(monitor = "valid_loss",
patience = 5)
)
)
preds <- predict(fitted, test_dl)
preds <- as.matrix(preds$to(device = "cpu"))
preds <- as.data.table(preds)
setnames(preds, names(preds), names(train_targets[, !"EntryID"]))
preds <- melt(preds)
preds_cv[, pred := pred + preds[, value] / 10] # mean for 10 iterations
return(NULL)
}
gc()
lapply(c(1:10), mlp_fit_predict_torch, num_epochs = 100)
gc()
preds_cv <- preds_cv[pred > 0.01]
preds_cv <- preds_cv[, .SD[order(-pred)][1:min(.N, 500)], by = .(EntryID)]
preds_cv[, pred := round(pred, 3)]
qsave(
preds_cv,
paste0("submits/tmp_mlp_upd_torch10_v3_tune2_p5_valid_on_train_",
aspect_name, "_by_freq", ".qs")
)
all_preds <- rbind(all_preds, preds_cv)
rm(preds_cv)
gc()
}
Sys.time() - t0
#--------------save all three aspects-------------------------------------------
qsave(
all_preds,
paste0("submits/esm2_torch10_top_by_freq_patience5_2755555", ".qs")
)
data.table(
"Unique proteins" = length(unique(all_preds$EntryID)),
"Unique terms" = length(unique(all_preds$term)),
"Min terms per prot" =
all_preds[, .(n_per_prot = .N), by = EntryID][, min(n_per_prot)],
"Max terms per prot" =
all_preds[, .(n_per_prot = .N), by = EntryID][, max(n_per_prot)],
"Number of predictions" = nrow(all_preds),
"Min." = round(min(all_preds[, pred]), 3),
"Median" = round(median(all_preds[, pred]), 3),
"Mean" = round(mean(all_preds[, pred]), 3),
"Max." = round(max(all_preds[, pred]), 3)
) |> t()Использовали оптимизатор AdamW, once cycle learning rate и раннюю остановку (early stopping callback) по значению функции потерь на валидации с patience = 5, то есть при отсутствии уменьшения значения функции потерь (binary cross entropy loss) после 5 эпох обучение останавливалось.
Похожие модели обучались также на tensorflow:
Обучение модели с использованием tensorflow
if (!dir.exists("submits")) dir.create("submits")
library(reticulate)
use_condaenv("tf")
library(tensorflow)
library(keras)
library(data.table)
library(qs)
library(seqinr)
min_freq <- c("BPO" = 250, "CCO" = 50, "MFO" = 50)
train_sequences_file <- "data_source/Train/train_sequences.fasta"
train_terms_file <- "data_source/Train/train_terms.tsv"
test_features <- qread("data_prepared/test_features_esm2_t36_3B.qs")
#--------------------- Prepare terms by sequence data --------------------------
# fasta_seq <- qread(train_sequences_file)
fasta_seq <- read.fasta(
train_sequences_file,
seqtype = "AA",
as.string = TRUE
)
fasta_seq <- data.table(
EntryID = names(fasta_seq),
fasta = unlist(fasta_seq)
)
# Load train targets
train_targets_by_seq <- fread(train_terms_file , sep = '\t')
# Remove subontologies roots
train_targets_by_seq <- train_targets_by_seq[
! term %chin% c("GO:0008150", "GO:0005575", "GO:0003674")
]
# Add sequences
train_targets_by_seq[fasta_seq, sequence := i.fasta, on = "EntryID"]
# Replace duplicated EntryIDs with first one for each sequence
train_targets_by_seq[, EntryID := .SD[1, EntryID], by = sequence]
# Remove duplicated annotations
train_targets_by_seq <- unique(train_targets_by_seq)
uniqueN(train_targets_by_seq$EntryID)
#------------------ Predict by ontology ----------------------------------------
t0 <- Sys.time()
all_preds <- NULL
for (aspect_name in c("BPO", "CCO", "MFO") ) {
freq <- min_freq[aspect_name]
train_all_targets <- train_targets_by_seq[aspect == aspect_name]
labels_to_consider <- train_all_targets[, .(n = .N), by = term]
labels_to_consider <- labels_to_consider[, .SD[order(-n)]]
labels_to_consider <- labels_to_consider[n > freq]
train_targets <- dcast(
train_all_targets[labels_to_consider, on = "term", nomatch = 0],
EntryID ~ term, length,
fill = 0
)
cols <- setdiff(names(train_targets), "EntryID")
train_targets[, (cols) := lapply(.SD, as.numeric), .SDcols = cols]
train_features <- qread("data_prepared/train_features_esm2_t36_3B.qs")
# Remove proteins without selected terms
train_features <- train_features[
train_targets[, EntryID],
on = "EntryID",
nomatch = 0
]
# 10-fold cross-validation
set.seed(42)
cv10 <- data.table(id = 1:train_features[, .N])
cv10[, folds := sample(1:10, .N, replace = TRUE)]
n_features <- ncol(train_features) - 1
n_targets <- ncol(train_targets) - 1
d_hid1 <- 2048
wd = 0.5
lr = 0.0002
bs = 256
dropout_prob = 0.5
num_epochs = 100
preds_cv <- data.table(
EntryID = rep(test_features$EntryID, times = n_targets),
term = rep(names(train_targets[, !"EntryID"]), each = test_features[, .N]),
pred = 0
)
mlp_fit_predict <- function(fold, num_epochs = 1) {
cat("\n--------------- ", aspect_name, " ", train_features[, .N],
" proteins; Fold number: ", fold, " ---------------\n", sep = "")
train_ids <- cv10[folds != fold, id]
valid_ids <- cv10[folds == fold, id]
#------------Standardization (Z-score normalization)-----------------------#
means <- sapply(train_features[train_ids, !"EntryID"], mean)
sds <- sapply(train_features[train_ids, !"EntryID"], sd)
cols <- names(train_features[, !"EntryID"])
train_f <- copy(train_features)
test_f <- copy(test_features)
for (col in cols) {
train_f[, (col) := (get(col) - means[col]) / sds[col]]
test_f[, (col) := (get(col) - means[col]) / sds[col]]
}
train_x <- as.matrix(train_f[train_ids, !"EntryID"])
train_y <- as.matrix(train_targets[train_ids, !"EntryID"])
validation_data <- list(
as.matrix(train_f[valid_ids, !"EntryID"]),
as.matrix(train_targets[valid_ids, !"EntryID"])
)
#---------------------model------------------------------------------------#
model <- keras_model_sequential(input_shape = n_features) %>%
layer_dense(d_hid1, activation = "relu") %>%
layer_dropout(rate = dropout_prob) %>%
layer_dense(d_hid1, activation = "relu") %>%
layer_dropout(rate = dropout_prob) %>%
layer_dense(n_targets, activation = "sigmoid")
model %>% compile(
optimizer = optimizer_adam(learning_rate = lr,
weight_decay = wd),
loss = "binary_crossentropy",
metrics = list(metric_auc())
)
callbacks <- list(
callback_early_stopping(patience = 5),
callback_progbar_logger(),
callback_reduce_lr_on_plateau(factor = 0.5,
patience = 1,
verbose = 1)
)
model %>%
fit(train_x,
train_y,
validation_data = validation_data,
batch_size = bs,
epochs = num_epochs,
callbacks = callbacks,
verbose = 2)
preds <- predict(
model,
as.matrix(test_f[, !"EntryID"])
)
preds <- as.data.table(preds)
setnames(preds, names(preds), names(train_targets[, !"EntryID"]))
preds <- melt(preds)
preds_cv[, pred := pred + preds[, value] / 10] # mean for 10 iterations
return(NULL)
}
gc()
lapply(c(1:10), mlp_fit_predict, num_epochs = 100)
gc()
preds_cv <- preds_cv[pred > 0.01]
preds_cv <- preds_cv[, .SD[order(-pred)][1:min(.N, 500)], by = .(EntryID)]
preds_cv[, pred := round(pred, 3)]
qsave(
preds_cv,
paste0("submits/tmp_mlp_upd_tf", aspect_name, "_by_freq", ".qs")
)
all_preds <- rbind(all_preds, preds_cv)
rm(preds_cv) ; gc()
}
Sys.time() - t0
qsave(
all_preds,
paste0("submits/esm2_tf_top_by_freq_patience5_2505050", ".qs")
)
data.table(
"Unique proteins" = length(unique(all_preds$EntryID)),
"Unique terms" = length(unique(all_preds$term)),
"Min terms per prot" =
all_preds[, .(n_per_prot = .N), by = EntryID][, min(n_per_prot)],
"Max terms per prot" =
all_preds[, .(n_per_prot = .N), by = EntryID][, max(n_per_prot)],
"Number of predictions" = nrow(all_preds),
"Min." = round(min(all_preds[, pred]), 3),
"Median" = round(median(all_preds[, pred]), 3),
"Mean" = round(mean(all_preds[, pred]), 3),
"Max." = round(max(all_preds[, pred]), 3)
) |> t()Небольшое примечание по поводу обучения на GPU/CPU. Код с использованием torch по умолчанию использует имеющуюся видеокарту. Для переключения расчетов на CPU достаточно указать accelerator = accelerator(cpu = TRUE) внутри вызова fit(). tensorflow можно заставить считать на CPU при помощи контекстного менеджера: вызовы функций для определения, компиляции и обучения модели нужно поместить внутрь with(tf$device("CPU"), {...}).
Постобработка
Полученные таким образом предсказания не являются консистентными. Дочерний терм может получить больший предикт (~большую вероятность наличия у белка), чем его предшественник в графе. Это неправильно, т.к. наличие дочернего терма по определению подразумевает наличие всех его родительских термов. В то же время включение корректировки предсказаний в саму процедуру обучения (как это сделано, например, в библиотеке SPROF-GO) также не является оптимальным подходом. Теоретическое обоснование состоит в том, что более специализированный терм может быть гораздо более легким для предсказания, чем его предки. Например, за ту или иную каталитическую активность отвечает вполне понятный участок фермента - активный центр. Хоть этот участок не обязательно образован непрерывным фрагментом первичной структуры, эмбеддинги языковой модели могут содержать адекватное числовое представление искомого фрагмента. Но если взять все ферменты в целом, то многообразие всех возможных активных центров слишком велико, чтобы на доступной выборке можно было научиться отделять все ферменты от не-ферментов. На практике так и оказалось, портированная из SPROF-GO функция потерь давала худшие результаты.
Поэтому для каждой модели выполнялась замена предсказаний всех термов на максимум предсказаний их дочерних термов:
Корректировка предиктов
library(data.table)
library(qs)
library(future)
library(furrr)
library(ontologyIndex)
future::plan("multisession", workers = 10)
file_name <- "submits/submit"
preds <- qread(paste0(file_name, ".qs"))
preds
preds <- unique(preds)
uniqueN(preds$EntryID)
# Load ontology
ontology <- get_ontology(
"data_source/Train/go-basic.obo",
propagate_relationships = c("is_a", "part_of")
)
# Add offspring terms to predictions
get_child_terms <- function(term, used_terms) {
result <- c(term)
while (! all(is.na(term))) {
term <- unlist(lapply(term, function(x) ontology$children[[x]]))
term <- unique(term)
term <- intersect(term, used_terms)
result <- c(result, term)
}
unique(result)
}
# Make a list with offspring of each term
dt <- data.table(term = preds[, unique(term)])
offspring_terms <- split(dt, dt[, term])
offspring_terms <- future_map(
offspring_terms,
function(term) {
unlist(get_child_terms(term, dt$term))
},
.progress = TRUE
)
preds[, pred_cor := NA_real_]
preds <- split(preds, preds[, EntryID])
rm(dt, ontology)
gc()
tictoc::tic()
preds <- future_map(
preds,
function(dt) {
res <- sapply(
dt$term,
function(y) dt[term %chin% offspring_terms[[y]], max(pred)]
)
dt[, pred_cor := res]
dt
},
.progress = TRUE
)
preds <- rbindlist(preds)
tictoc::toc()
preds[pred != pred_cor, .N]
qsave(preds, paste0(file_name, "_corr.qs"))
compute_stats <- function(dt) {
data.table(
"Unique proteins" = length(unique(dt$EntryID)),
"Unique terms" = length(unique(dt$term)),
"Min terms per prot" =
dt[, .(n_per_prot = .N), by = EntryID][, min(n_per_prot)],
"Max terms per prot" =
dt[, .(n_per_prot = .N), by = EntryID][, max(n_per_prot)],
"Number of predictions" = nrow(dt),
"Min." = round(min(dt[, pred]), 3),
"Median" = round(median(dt[, pred]), 3),
"Mean" = round(mean(dt[, pred]), 3),
"Max." = round(max(dt[, pred]), 3)
)
}
compute_stats(preds)Это очень вычислительно затратный фрагмент кода, которые много раз переписывался для улучшения быстродействия и снижения требований по памяти.
Ансамблирование и снова постобработка
Скорректированные предикты выбранных 6 моделей усредняли и еще раз проводили корректировку, т.к. после усреднения снова возможно образование коллизий. Затем предикты топ450 по частоте термов оставляли как есть, а из остальных оставляли только те, которые больше 0.5.
Дальше делали простую, но не очевидную процедуру: брали все доступные неэкспериментальные аннотации, присваивали им предикт, равный 1, и усредняли предсказания ансамбля нейросеток с тем, что получилось. Это и был первый выбранный для зачета сабмит. Второй - все то же самое, но вместо последнего усреднения использовались взвешенная сумма и дополнительно отбиралось топ35 предсказаний на белок (результаты на публичном лидерборде почти не отличались).
Был еще один этап постобработки, не представляющий большого интереса и образовательной ценности. В этом посте кода не будет, он выложен тут. На некоторые термы существуют ограничения по таксонам. Так, например, связанные с ядром функции отсутствуют у прокариот, и такие абсурдные предсказания желательно удалять. К сожалению, учет этих ограничений никак не повлиял на результат.
Результаты
Если речь не идет о призовых (денежных) местах, то полученное на лидеборде место представляет собой самую малую часть пользы от участия в соревновании.
В данном случае:
взяли золото;
получилось не шейкапнуться вниз - 16 место на публичном лидерборде, 17 на промежуточном приватном и финальный результат в виде 13 места;
почти не оверфитнулись, несмотря на не слишком тщательную локальную валидацию (хоть качество и просело до ~0.55 по сравнению с ~0.61 на публичном лидерборде, почти у всех участников в топе наблюдалась похожая картина; более-менее “при своих” остались первые два места).
С технической точки зрения итоговое решение удалось реализовать средствами R, хотя без некоторого количества кода на питоне в процессе экспериментов не обошлось. Основной “рабочей лошадкой” был torch версий 0.10 и 0.11 и пакет luz, реализующий высокоуровневый API к нему. Также использовали tensorflow v2.12 посредством reticulate. Код работает как на Linux, так и на Windows. Вычислительные ресурсы были довольно скромными: 1 ПК с CPU на 8 ядер, 32 Гб ОЗУ и GTX1070 и 1 ПК с CPU на 8 ядер, 128 Гб ОЗУ и RTX3090.
Важным наблюдением была неполная воспроизводимость результатов между разными версиями torch-а (0.10 и 0.11), а также для одних и тех же нейросеток, обученных на GPU и CPU. Перенос с Linux на Windows и наоборот тоже иногда преподносит сюрпризы. Какой-то “правильной” конфигурации выбрать не удалось, обученные на разных конфигурациях сетки были включены в итоговый ансамбль.
Многие использованные приемы весьма универсальны и могут быть полезны при решении типовых реальных задачах: обработка табличных данных с использовнием data.table, хранение результатов в формате qs, параллелизация при помощи future, перенос матричных вычислений на сторону torch с задействованием GPU и некоторые другие моменты, которые можно видеть в представленных фрагментах кода.
Лишний раз повторю банальные советы, в справедливости которых снова убедился:
тщательно валидируйтесь локально по правильной метрике и верьте прежде всего своей валидации, а не лидерборду (тем более что за пределами соревновательных площадок никаких лидербордов нет);
критически рассматривайте любые опубликованные решения,
даже еслиособенно если они дают хороший результат на лидерборде;не спешите участвовать с самого первого дня соревнования.
Наконец, главные методологические уроки, извлеченные в процессе.
Все повторяющиеся действия были своевременно оформлены в функции с относительно понятными интерфейсами, но код все же нужно дополнительно структурировать и документировать. Как вариант, оформлять в виде пакета и активнее использовать контроль версий.
В команде из 2 человек получилось обойтись обменом файлов через телеграм, но лучше было бы работать с приватным репозиторием на github/gitlab. Имела место классическая недооценка сложности проекта. Также для организации работы в команде большего размера пригодился был slack и/или группа в телеграме с топиками, а может быть и сервисы типа trello.
Для серьезной борьбы за призовые места лучше собирать команду из участников с разнообразными бэграундами и программистскими стэками. Мы все сделали на R скорее в качестве proof of concept и для прокачки навыков, чем по реальной необходимости. Решения некоторых задач получились удачными по сравнению с опубликованными версиями на питоне, но для их реализации пришлось интенсивно гуглить, писать и тестировать много вариантов кода вместо того, чтобы не тратить время и скопировать готовое. За пределами соревновательных площадок обычно нужно находить компромисс между этими крайностями.
Комментариев нет:
Отправить комментарий